DSB break processing and repair in the DNA damage response
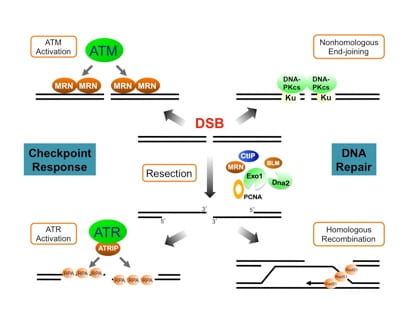
The DNA damage response (DDR) induced by double-strand breaks (DSBs) is a highly sophisticated system that safeguards the integrity and stability of the genome. Our work in DDR focuses on understanding a key process called DNA end resection, which dictates the overall mode of the damage response. By converting DSB ends into ssDNA structure, DNA resection promotes the activation of the ATR-dependent checkpoint and attenuates the ATM-mediated checkpoint. Resection also channels DSBs into homology-mediated repair and averts repair by nonhomologous end-joining. We have identified the tumor suppressor protein CtIP as a key player in linking damage sensing to end resection after DSB damage. Moreover, we have provided evidence that cells deploy distinct factors and strategies when initiating resection at “clean” DSBs (those with free ends) and “dirty” DSBs (those with chemical or protein adducts). Furthermore, our work on the resection extension and termination processes has provided crucial insights into how the Exo1 resection pathway is tightly regulated by multiple factors to ensure a proper level of resection that is essential for genome stability. We are extending our studies to further unravel the molecular mechanisms of the DSB resection process and its regulation by checkpoint pathways and other factors.
A novel cytosolic DNA/Ca2+-dependent signaling pathway for genome protection during replication stress
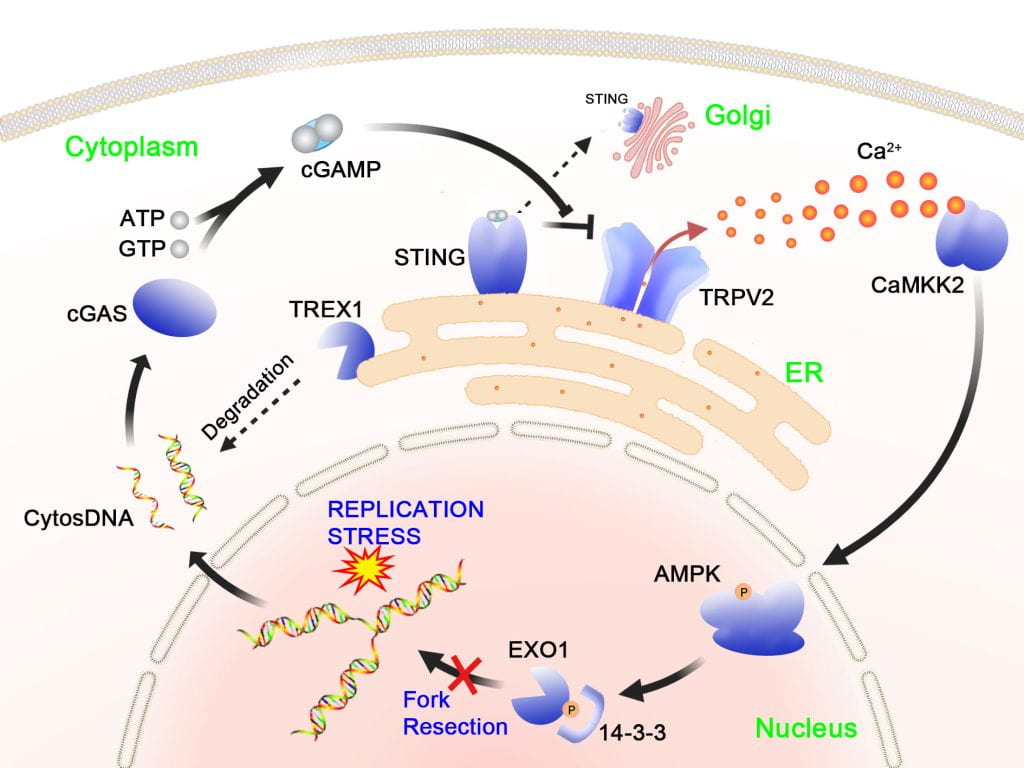
The replication stress response (RSR) operates in S phase to protect the genome and to ensure complete DNA replication. Defects in replication controls are a major source of cancer-associated mutations and genomic instability. We have recently identified a novel signaling pathway that defends replication forks from aberrant processing upon replication stress. In this pathway, replication stress, which can be induced by cancer drugs or oncogene activation, gives rise to cytosolic self-DNA, which in turn activates cGAS, leading to cGAMP production. The binding of cGAMP to STING causes its dissociation from the ion channel TRPV2 on the ER, leading to TRPV2 derepression and Ca2+ release. The resulting elevation of Ca2+ activates CaMKK2 and a downstream kinase AMPK. Following activation, AMPK directly phosphorylates the EXO1 nuclease at serine-746, leading to binding of 14-3-3 proteins and inhibition of fork association of EXO1. As a result, abnormal fork resection is avoided. Disruption of this pathway causes chromosomal instability and reduced cell viability. Interestingly, this calcium-mediated pathway functions separately from the replication checkpoint pathway, which also protects fork structure.
Our ongoing research is directed to further decipher this pathway, focusing on the physical and function al interaction between STING and TRPV2 and the mechanisms of cytosolic DNA production and its translocation into the cytosol. We are also exploring new functions of this signaling pathway in other cellular processes such as innate immune response, autophagy and metabolic control.
Regulation of NMD by intracellular Ca2+
Like DNA, RNA in the cell is also constantly monitored for quality control. Nonsense-mediated mRNA decay (NMD) is a RNA surveillance pathway that selectively degrades transcripts bearing premature translation termination codons (PTCs), thereby preventing the synthesis of potentially harmful protein products. This pathway also serves as a gene regulatory mechanism that controls the expression levels of numerous physiological mRNAs. NMD has recently emerged as a potent modulator of tumorigenesis and as an attractive target for cancer intervention.
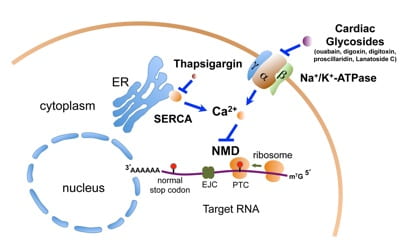
To understand the NMD pathway and to develop NMD therapeutics, we have developed a dual-color, bioluminescence-based reporter system that can effectively measure NMD activity in live human cells. In a high-throughput screen using this reporter system, we identified a group of cardiac glycosides (CGs) such as ouabain and digitoxin as potent inhibitors of NMD. These CGs inhibit NMD through their binding to and inhibition of the Na+/K+-ATPase on the plasma membrane. Our further studies have led to the discovery that intracellular calcium is a key regulator of NMD, with an increase in calcium levels suppressing NMD. This finding suggests that NMD is a highly dynamic process and that NMD is part of the gene expression programs controlled by calcium. Building on these findings, we are currently investigating the molecular basis of the calcium-mediated NMD regulatory pathway and exploring the physiological significance of this novel regulation in various biological contexts.
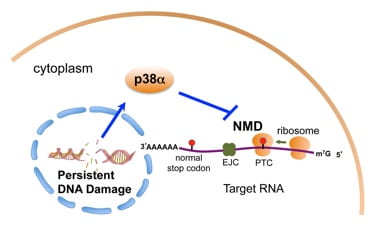
NMD in the cellular response to persistent DNA damage
The multi-step gene expression process involves the formation, maturation and remodeling of various ribonucleoprotein (RNP) complexes. Mechanistic coupling of these steps and active surveillance on the RNP complexes ensure efficient and faithful gene expression. Importantly, emerging evidence suggests that these coupling and surveillance mechanisms also prevent the formation of harmful structures such as R-loops (RNA:DNA hybrid) that could cause DNA damage and genomic instability. Conversely, the DDR also regulates gene expression by impinging on transcription, splicing and translation, and likely also on RNA surveillance pathways.
We have recently found that persistent DNA damage in non-cycling cells leads to inhibition of the NMD RNA surveillance/gene regulation pathway. p38a MAPK is a key mediator of this NMD inhibition, and this regulation apparently plays an important role in the reprogramming of gene expression in cells with chronic DNA damage. The impact of persistent DNA damage on NMD in non-cycling cells may be relevant to the remodeling of the tumor microenvironment following radiotherapy or chemotherapy. We continue to dissect the molecular basis and physiological significance of the DDR-NMD link. In addition, we are investigating the roles of NMD factors in genome maintenance in the absence of exogenous DNA damage.